Alois Alzheimer reported lipid-filled glia surrounding amyloid plaques in the AD brain. But how and why they appear remains elusive. Now, two bioRxiv preprints blame Aβ. In one, posted June 6, scientists led by Gaurav Chopra of Purdue University, West Lafayette, Indiana, and Dimitrios Davalos at Ohio’s Cleveland Clinic reported that mouse microglia shift their lipid metabolism when exposed to Aβ fibrils in vitro. The cells form lipid droplets, courtesy of overzealous diglycerol acetyltransferase, aka DGAT2, an enzyme that creates triglycerides from free fatty acids. These droplet-filled microglia poorly phagocytosed Aβ. In the other manuscript, uploaded July 25, scientists led by Tony Wyss-Coray, Stanford University, described Aβ-induced triglyceride droplets in human microglia and attributed them to an uptick in ACSL1, an enzyme upstream of DGAT2.
APOE4 exacerbated lipid droplet formation in these cells, and lipid-filled microglia dotted hippocampal tissue from APOE4/4 carriers. The glia also exuded something neurotoxic, perhaps the lipids themselves, hiking up phospho-tau and programmed cell-death proteins produced by nearby neurons.
“This work brings into sharp focus the mechanisms underlying one of Alzheimer’s original observations, that lipids accumulate in microglia, which has received relatively scant attention to date,” wrote Anne Poljak, University of New South Wales in Sydney. Priyanka Narayan at the National Institute of Diabetes and Digestive and Kidney Diseases in Bethesda, Maryland, noted how far this research area has come over the past decade. “It is clear that lipids in neurodegenerative disease have entered a new era of importance; however, there is still a long way to go to understand the mechanisms that govern their contribution to different cell types in initiation and progression of the disease,” she wrote (comments below).
Lipids and Droplets. ACSL1 adds acetyl-CoA onto free fatty acids (FFA) to form Acyl-CoAs. Thioesterases (Them1 and Them2) can reverse the process, but Glycerol-3-phosphate acyl transferase (GPAT) and DGAT2 (not shown) can combine these acyl-CoAs to form triacylglycerols, aka triglycerides. TGs typically bind very low-density lipoproteins (VLDL), but they can also form lipid droplets (yellow circle). (Courtesy of Desai et al., 2018).
Microglia and macrophages make lipid droplets when stressed by inflammation. Scientists believe the lipids sustain the cells’ metabolic need, enabling them to respond to threats (van Dierendonck et al., 2022; reviewed by Olzmann and Carvalho, 2019). Wyss-Coray previously described lipid-droplet accumulating microglia (LDAMs) that spewed pro-inflammatory cytokines in the hippocampi of old wild-type mice (Aug 2019 news). Matthew Blurton-Jones, University of California, Irvine, found that human microglia placed into the brain of an amyloidosis mouse filled up with lipid droplets (Claes et al., 2021).
Could Aβ spur LDAM formation? Both research groups suggest as much. In vitro, an hour after adding synthetic Aβ42 fibrils to cultured wild-type mouse microglia, co-first authors Priya Prakash and Palak Manchanda in Chopra’s lab saw the lipid droplet content swell 4.5-fold. Co-first authors Michael Haney and Róbert Pálovics in Wyss-Coray’s lab saw a similar uptick in human induced pluripotent stem cell-derived microglia after 24 hours of Aβ42 fibril exposure.
In vivo, the teams spotted LDAMs near amyloid plaques in 6-month-old 5xFAD mice, and saw similar cells in hippocampal tissue from people who had had AD (see image below). In each case, the cells were filled with lipid droplets. Notably, Aβ fibrils provoked no lipid droplet formation in cultured 5xFAD microglia, perhaps, Davalos suggested, because they were already stuffed with lipids.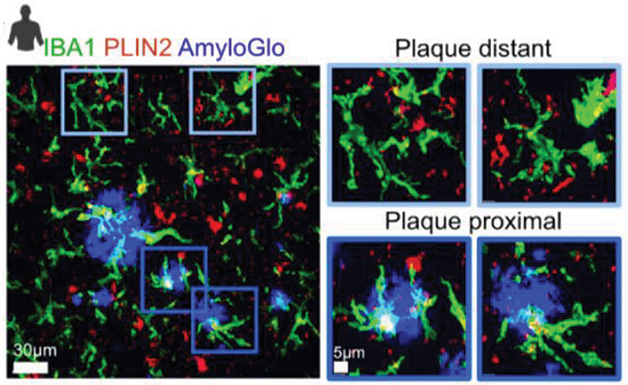
White Fat. High-resolution confocal microscopy of AD brain shows microglia (green) near amyloid plaques (blue) brimming with lipid droplets (red) that appear white in overlay images. Microglia far away were lipid-free (top). [Courtesy of Prakash et al., bioRxiv, 2023.]
Does Aβ change the microglial lipidome? Prakash reported that, after 24 hours with the fibrils, wild-type microglia contained few fatty acids but an abundance of triglycerides, mirroring the lipidome of 5xFAD LDAMs. Haney saw the same rise in triglycerides. These fats, along with cholesterol esters, constitute the core of lipid droplets, connecting the lipidomic and phenotypic changes (Listenberger et al., 2003; Fujimoto and Parton, 2011). Chopra thinks this ramped-up triglyceride synthesis could be the microglia’s way of controlling a flood of free fatty acids, which can easily be oxidized to become toxic.
What caused this shift? Both groups suspected enzymes in the triglyceride synthesis pathway. Wyss-Coray pegged changes to long-chain-fatty-acid—CoA ligase 1 (ACSL1), which tacks on CoA to free fatty acids. Chopra pegged diacylglycerol O-acyltransferase 2 (DGAT2), which turns the fatty-acyl-CoAs into triglycerides. ACSL1 overexpression induces lipid droplet formation in multiple cell types; DGAT2 flocks to lipid droplets, where it makes triglycerides (Zhao et al., 2020; Kuerschner et al., 2008).
ACSL1 is implicated in ferroptosis. Instigated by oxidized lipids, this cell death pathway is linked to Alzheimer’s, Parkinson’s, and Huntington’s diseases (Bai et al., 2019; reviewed by Reichert et al., 2020).
In single-nucleus RNA-Sequencing analysis of AD cortical tissue, Haney et al. saw a subset of ASCL1-expressing LDAMs surrounding amyloid plaques. ASCL1 expression rose hand-in-hand with lipid droplet formation, indeed it was the most upregulated gene in microglia from AD cases compared to cells from controls. Likewise, Prakash et al. spotted DGAT2-positive LDAMs surrounding plaques in immunostained mouse and human brain tissue (image below). Both groups concluded that those enzymes likely drove lipid droplet formation.
Droplets and DGAT2. In AD hippocampal tissue (bottom), microglia (green) near amyloid plaques (blue) contained lipid droplets (pink) and DGAT2 (yellow). [Courtesy of Prakash et al., bioRxiv, 2023.]
The scientists next asked if APOE4 greases this process, since this lipoprotein strongly binds triglyceride-rich lipoprotein particles (Mahley, 2016). Indeed, they found that APOE4 homozygotes who had AD had more LDAMs in their brains than did APOE3 homozygotes with AD, and they expressed more ASCL1 in their microglia. APOE4 fit this picture in vitro, too. Human iPSC-derived APOE3/3 microglia had few lipid droplets with or without exposure to Aβ fibrils, whereas E4/4 cells had some at baseline, generated many more in the presence of Aβ, and ramped up ACSL1 expression.
Does the Fat Matter?
Lipid-laden APOE4/4 microglia seemed to poison neurons. Human iPSC-derived neurons bathed in media from E4/4 LDAMs expressed the apoptosis indicator caspase, made AT8-positive phospho-tau, and contained lipid droplets made of the same triglyceride species found in droplets from LDAMs. To Haney, this suggests that the LDAMs secrete these triglycerides, which nearby neurons take up.
Death by Fat? When microglia sense Aβ fibrils, among other inflammatory triggers, they upregulate ACSL1, creating an abundance of triglycerides that form lipid droplets with the help of ApoE. These LDAMs (red) secrete molecules, perhaps the lipids themselves, that are toxic to neurons. [Courtesy of Haney et al., bioRxiv, 2023.]
On the flip side, consider APOE knockout microglia. Aβ fibrils did not provoke lipid droplets or ACSL1 production in them, and the conditioned medium was harmless to neurons. To Julia TCW of Boston University, these results add weight to the notion that the APOE4 allele has a toxic gain of function (comment below).
Sarah Cohen at the University of North Carolina, Chapel Hill, agrees. In her lab’s preprint, uploaded in April 2023, Cohen saw ApoE4 astrocytes bloat with bubbles of triglycerides and found ApoE4 interacting with lipid droplets, attempting to control their size. The protein fumbled this lipid turnover, allowing large droplets to form (Windham et al., 2023). Cohen suspects the same happens in microglia exposed to Aβ. “Our data indicate that APOE plays a previously unrecognized role as a [lipid droplet] surface protein that regulates size and composition,” wrote Cohen and colleagues in their manuscript.
Is There a Drug Target in There?
Knocking down ACSL1 or DGAT2 might dissolve lipid droplets, though perhaps with unintended consequences. Previously, Li-Huei Tsai and Matheus Victor at MIT reported that blocking ACSL1 for two days killed microglia. “Lipid accumulation influences many processes and must be fine-tuned rather than abrogated,” wrote Tsai and Victor (comment below).
For their part, Prakash and colleagues saw, after applying a DGAT2 inhibitor for two hours to cultured 5xFAD microglia, a halving of their lipid droplet number and more Aβ phagocytosis. In 2-year-old 5xFAD mice, a week-long intraventricular infusion of a molecule that degrades DGAT2 cut LDAMs by one-third and amyloid plaques by 60 percent. Chopra was tight-lipped about this molecule, only saying that it is a bifunctional, small-molecule conjugate. He plans to upload a preprint on it to bioRxiv soon, he told Alzforum.
DGAT2 inhibitors are already being tested in people. Pfizer’s small-molecule drug ervogastat and Ionis’s ASO ION224 are both in Phase 2 trials for non-alcoholic steatohepatitis, a form of fatty liver disease that can cause the organ to fail (Amin et al., 2022; clinical trials.gov).
Others shared Tsai and Victor’s concern. Jörg Hanrieder, University of Gothenburg, said these lipid processing enzymes are crucial throughout the body. Ole Isacson of McLean Hospital, Belmont, Massachusetts, stressed that microglia need lipid droplets to initiate an immune response (Bosch et al., 2020). Edoardo Marcora, Icahn School of Medicine, Mount Sinai, New York, thinks lipid droplets may not be the “bad guy.” “Lipid droplets might just be a beneficial adaptive response to something that causes dyshomeostasis, and this must be understood before targeting them with drugs,” he said.—Chelsea Weidman Burke
Haney et al. and Prakash et al. focus on a little-studied attribute of the AD brain, i.e., the accumulation of lipids in glia, and their potential mechanistic role in neuronal cell death. As a rationale for their work, Haney et al. cite Alois Alzheimer’s original histopathologic observation that “many glial cells show adipose saccules.” Prakash et al. point out that the hallmark AD feature (Aβ accumulation in plaque) has had setbacks as a sole explanation of cognitive decline in AD, and point to “reactive microglia [which] cluster around Aβ plaques” as a point of interest.
Both groups then characterize a subset of microglia that accumulate LDs, and which associate in proximity with Aβ plaques, with Prakash et al. using the 5xFAD mouse model together with human brain tissue, while Haney et al. use human AD postmortem tissue. Using transcriptomics, and a comparison of AD brains, Haney showed higher expression of the ACSL1 gene in AD versus age-matched controls, and in AD APOE4 homozygotes versus AD APOE3 homozygotes. Using immunohistochemistry, they also show that not only ACSL1 gene expression, but also the ACSL1 protein, is upregulated in AD brain, and more so in APOE4 than in APOE3 carriers. ACSL1 expression was specific to “lipid droplet accumulating microglia (LDAM)” which they define as a distinct microglial state. As ACSL1 is a regulator of lipid metabolism, Haney et al. suggest that it is responsible for increased microglial lipid droplet accumulation and triglyceride synthesis.
Prakash et al. show that LD-rich microglia are associated in proximity to Aβ plaques in both the mouse model and in human brain tissue. Using lipidomic and metabolomic profiling, they identify changes to a diversity of lipids and metabolic pathways, which they suggest account for the LD-laden microglial phenotype. They show a decrease in free fatty acids together with an increase in triglycerides. They attribute this to the enzyme DGAT2, which converts free fatty acids to triglycerides.
Furthermore, Prakash et al. identified an interesting conundrum—that direct exposure of microglia from wild-type mice to a fluorescent-labelled Aβ (AβpH) increased LD content by 4.5-fold, but microglia from 5xFAD mice had no significant LD increase.
While the two groups use contrasting study designs, both contain a rich array of observational and functional experiments, showing dysregulation of lipid metabolism, increased TG levels, and changes to the expression of lipid regulating genes and proteins in AD, which likely account for the LDAM phenotype. Both relate the formation of LD-rich microglia to the effects of aberrant Aβ forms; fibrillar Aβ (Haney et al.) and Aβ1-42 (Prakash et al.). Interestingly, each team comes to somewhat different conclusions about the neuropathological implications of the LDAM phenotype. Prakash et al. suggest that LD-rich microglia are impaired in their ability to uptake the toxic Aβ1-42 variant, while Haney et al. suggest that LD accumulation in glia leads to glial secretion of neurotoxic factors.
In aggregate, these findings bring into sharp focus the mechanisms underlying Alzheimer’s original observation of lipid accumulation in microglia, which has had relatively scant attention to date. Both show a dysregulation of lipid metabolism; this is likely due to enzymes involved, such as ACSL1 and DGAT2, but likely also other enzymes responsible for lipid expression and turnover.
These studies open the door for additional work to address questions such as:
(a) Specifically, which forms of Aβ can cause microglial AD accumulation? The current work suggests fibrillar Aβ and Aβ1-42, but what about Aβ1-40, which can also aggregate (Vander Zanden et al., 2019) and is the predominant version in cerebral amyloid angiopathy. Or what about monomeric versus fibrillar Aβ forms, or indeed amyloid plaque, which has sometimes been considered an inert endpoint of AD?
(b) How do LDAMs contribute to neuropathology? Is it by increasing toxicity, or by decreasing the ability of microglia to protect from toxic endogenous Aβ variants, or by some combination of both?
(c) Are LDAMs an intermediate in the development and progression of AD, or could aberrant lipid metabolism be an AD trigger?
(d) How early in the development and progression of AD do changes to lipid metabolism occur, and are they evident in both familial and idiopathic AD variants?
(e) Is there an upper threshold of LD accumulation in cells, beyond which additional accumulation is not possible?
(f) What are the specific mechanisms by which LDAM may affect neuronal integrity?
(g) Lastly, that both AD homozygous APOE4 and APOE3 carriers have altered lipid metabolism relative to non-AD controls suggests that both genetic and lifestyle/environmental determinants can contribute to the LDAM phenotype. Non-genetic determinants suggest that there may be potential therapeutic or preventative options to be explored.
Vander Zanden CM, Wampler L, Bowers I, Watkins EB, Majewski J, Chi EY. Fibrillar and Nonfibrillar Amyloid Beta Structures Drive Two Modes of Membrane-Mediated Toxicity. Langmuir. 2019 Dec 3;35(48):16024-16036. Epub 2019 Sep 26 PubMed.
These two studies make it abundantly clear that lipid droplets modulate microglial function in AD models. Over the past decade, the research community has rediscovered the importance of lipid droplet pathology in AD, and has made considerable strides in delineating the mediators, modulators, and effects of these previously overlooked organelles.
These preprints use an array of model systems—immortalized cell lines, human AD tissue, mouse models, human iPSC-derived microglia, mouse primary microglia. Haney and Palovics et al. focus on the role of human APOE isoforms, especially E4, on microglial LDs, whereas Prakash and Manchanda et al. use the familial AD mouse model 5xFAD to explore microglial LDs.
Data from all these models across the two manuscripts converge on a key finding: that Aβ exposure induces LD formation in microglia, and that these lipid-laden microglia have an array of detrimental phenotypes. These range from proinflammatory states to reduced phagocytosis and damaged lysosomes, phenotypes that have been observed in other studies as well (Lee et al., 2023). Aβ-induced LDs occur in the context of LOAD (as modeled by APOE4) as well as in the context of familial AD (modeled by 5xFAD), providing another bridge (in addition to amyloid and tau) between the late-onset and inherited forms of the disease.
For lipid afficionados, both preprints observe an increase of unsaturated triacylglycerides after 24 hours of Aβ exposure. This trend has been observed in multiple other contexts (Sienski et al., 2021; Windham et al., 2023). Prakash, Manchanda et al. explore the dynamics of this response by comparing acute (one hour) versus chronic exposure (24 hours); they observe a time-dependent change in both the saturation of fatty acids and storage of fatty acids in more complex lipids like TAGs and cholesterol esters. Haney and Palovics et al. focus on the 24-hour treatment paradigm. It is worth noting that the dose of amyloid used in Haney and Palovics et al. (5 μM) is 10 times that used in Prakash and Manchanda et al.
Both papers use various methods to identify modulators of the lipid droplet. Haney and Palovics et al. use single-cell transcriptomics from human brains and iPSC-derived microglia as well as CRISPRi screens in monocyte cell lines and iPSC-derived microglia. These screens reveal two methods for modulating lipid droplet formation—decreasing ACSL1 (a fatty acid acylation enzyme) or increasing lipolysis (by broadly inhibiting PI3K signaling)—though it should be noted that these enzymes feed into other anabolic and catabolic pathways as well. Prakash and Manchanda et al. focus on DGAT2—which catalyzes the rate-limiting step in TAG synthesis—inhibition of which alleviates some aspects of microglial dysfunction. This effect seems to be dependent on the AD context, suggesting that other lipid modulators may also play a role.
Both papers discover LD-containing microglia in the proximity of amyloid plaques, suggesting that these cells may have a role in responding, phagocytosing, or managing the growth and toxicity of plaques. Haney and Palovics et al. delve further into mechanisms of cell non-autonomous action. They report that conditioned media from microglia with high LD content can induce tau phosphorylation in iPSC-derived neurons. This builds on previous findings that aberrant lipid accumulation in APOE4 microglia can influence neuronal signaling (Victor et al., 2022).
After reading these preprints, I’d like to share these points with the community:
Together, it is clear lipids in neurodegenerative disease have entered a new era of importance. However, there is still a long way to go to understand the mechanisms that govern their contribution to different cell types in the initiation and progression of the disease.
Lee S, Devanney NA, Golden LR, Smith CT, Schwartz JL, Walsh AE, Clarke HA, Goulding DS, Allenger EJ, Morillo-Segovia G, Friday CM, Gorman AA, Hawkinson TR, MacLean SM, Williams HC, Sun RC, Morganti JM, Johnson LA. APOE modulates microglial immunometabolism in response to age, amyloid pathology, and inflammatory challenge. Cell Rep. 2023 Mar 28;42(3):112196. Epub 2023 Mar 3 PubMed.
Sienski G, Narayan P, Bonner JM, Kory N, Boland S, Arczewska AA, Ralvenius WT, Akay L, Lockshin E, He L, Milo B, Graziosi A, Baru V, Lewis CA, Kellis M, Sabatini DM, Tsai LH, Lindquist S. APOE4 disrupts intracellular lipid homeostasis in human iPSC-derived glia. Sci Transl Med. 2021 Mar 3;13(583) PubMed.
Windham IA, Ragusa JV, Wallace ED, Wagner CH, White KK, Cohen S. APOE traffics to astrocyte lipid droplets and modulates triglyceride saturation and droplet size. 2023 Apr 29 10.1101/2023.04.28.538740 (version 1) bioRxiv.
Victor MB, Leary N, Luna X, Meharena HS, Scannail AN, Bozzelli PL, Samaan G, Murdock MH, von Maydell D, Effenberger AH, Cerit O, Wen HL, Liu L, Welch G, Bonner M, Tsai LH. Lipid accumulation induced by APOE4 impairs microglial surveillance of neuronal-network activity. Cell Stem Cell. 2022 Aug 4;29(8):1197-1212.e8. PubMed.
van der Kant R, Langness VF, Herrera CM, Williams DA, Fong LK, Leestemaker Y, Steenvoorden E, Rynearson KD, Brouwers JF, Helms JB, Ovaa H, Giera M, Wagner SL, Bang AG, Goldstein LS. Cholesterol Metabolism Is a Druggable Axis that Independently Regulates Tau and Amyloid-β in iPSC-Derived Alzheimer’s Disease Neurons. Cell Stem Cell. 2019 Mar 7;24(3):363-375.e9. Epub 2019 Jan 24 PubMed.
Wyss-Coray and colleagues have identified a key regulator of lipid droplet (LD) biogenesis, ASCL1, activated specifically in microglia and in APOE4 human AD brain, then followed by triacylglycerol (TAG) synthesis and lipid droplet (LD) accumulation.
In vitro, iPSC-derived APOE4 microglia continually accumulate LDs upon challenge with fibrils of Aβ. Interesting results from the study are that conditioned media from high-LD APOE4 microglia induce tau phosphorylation and neurotoxicity in neurons but not conditioned media from low-LD APOE4 microglia nor, tellingly, from high-LD APOE knockout microglia. This demonstrates that APOE4 has a toxic gain of function, regardless of the presence of high LD in microglia.
Davalos, Chopra, and colleague have identified another key regulator of LD biogenesis in 5xFAD mice, DGAT2, also a part of the LD biogenesis pathway converting free fatty acids to TAGs. An interesting finding in this paper is that LD-laden microglia are in very close proximity to amyloid plaques, almost directly contacting Aβ, leading to a phagocytic defect. These authors further demonstrated that amyloid exposure is sufficient to induce microglial LD formation. Interestingly, a DGAT2 inhibitor does not reduce LD formation in microglia from 5xFAD upon Aβ exposure, but increases Aβ phagocytosis.
In short, these studies found, in common, that both APOE4 and amyloid activate key regulators of LD biogenesis in microglia, revealing potential targets for AD therapeutic intervention.
This exciting pair of preprints explores the role of microglial lipid droplet accumulation in Alzheimer’s disease (AD) using complementary human and rodent model systems. Haney used AD brain tissue and human stem cell-derived microglia (iMG) and neurons, while Prakash, Chopra, and colleagues primarily used a mouse model of AD. Both found that microglia from AD brains had altered lipid metabolic profiles, with triglycerides accumulating in lipid droplets, especially in microglia close to amyloid plaques.
Intriguingly, Haney et al. found that treating microglia with fibrillar Aβ induced lipid droplet formation in a genotype-dependent manner. APOE is a lipid-related AD risk gene, but how APOE genotype intersects with Aβ and tau to increase AD risk is unclear. Microglia expressing APOE4 accumulated more LDs in response to Aβ. Remarkably, these LD-high microglia released factors that induced hallmarks of AD pathology in cultured neurons. Media from LD-high microglia caused neurons to accumulate phosphorylated tau, while media from LD-low microglia did not. These experiments suggest that microglial APOE may act after Aβ and before tau in an amyloid-cascade model of AD.
These experiments raise many questions about how Aβ causes lipid droplet accumulation in microglia, and how this accumulation, in turn, causes release of factors that induce tau pathology within neurons. The studies each identified enzymes involved in triglyceride synthesis as key players. Haney et al. identified expression of ACSL1 as the key feature of plaque-associated microglia, while Prakash et al. showed that DGAT2 was required for microglial lipid droplet accumulation. ACSL1 converts fatty acids into fatty acyl-CoAs. DGAT2 uses diacylglycerol and a fatty acyl-CoA to synthesize triglyceride. Thus, both these enzymes work at the last step of triglyceride synthesis.
Excitingly, the two studies showed that reducing lipid droplets by inhibiting either PI3 kinase, or DGAT2, rescued multiple microglial phenotypes. Thus, this work opens new possibilities, suggesting that inhibiting triglyceride synthesis in microglia could be beneficial as part of a therapeutic strategy for AD.
My lab has had a continued interest in the cell-type-dependent impact of APOE4 expression across the brain, particularly as it relates to lipid metabolism in glia. We have previously created isogenic iPSC lines bearing either the disease-neutral allele APOE3, or the disease-associated allele APOE4, for phenotypic analysis (Lin et al., 2018). After deriving all major brain cell types from multiple APOE isogenic pairs, our study reported, amongst its many findings, that APOE4 astrocytes accumulated intracellular lipids, while APOE4 microglia exhibited hallmarks of inflammation and reduced Aβ phagocytosis (Lin et al., 2018). This led us to perform an in-depth characterization of lipid homeostasis in APOE4-bearing astrocytes, where we uncovered increased accumulation of unsaturated fatty acids and intracellular lipid droplets, both in APOE4-expressing human astrocytes and in an APOE4 yeast model (Sienski et al., 2021). Our study also demonstrated that exogenous choline supplementation was sufficient to reverse the APOE4-induced lipid defects.
We have recently examined the consequences of APOE4 expression in iPS-derived microglia in crosstalk with neurons. We found that lipid-droplet accumulation is dramatically increased in APOE4 microglia, and that this disrupted microglial surveillance of neuronal secreted cues (Victor et al., 2022). In our study, we identified a metabolic switch in APOE4 that favored glycolysis instead of mitochondrial respiration and that primed these cells toward a pro-inflammatory state. Interestingly, we also found that lipid-burdened APOE4 microglia were marked by the upregulation of ACSL1 and that depleting lipid droplets with an ACSL1-specific inhibitor could restore microglia surveillance of neuronal activity. From RNA-Seq analysis, we found little overlap between the APOE4-induced state with the previously reported lipid droplet accumulating microglia (LDAM) profile. Phenotypically, LDAM exhibits reduced phagocytosis and increased secretion of pro-inflammatory cytokines, which is similar to the cellular state we have previously characterized in APOE4 bearing iPSC-derived microglia (Lin et al., 2018). But contrary to our findings with human APOE4 iMGLs, mouse LDAMs exhibit increased mitochondrial respiration. The poor convergence of transcriptional signatures between our human APOE4 iMGL and mouse LDAM may be due to a transcriptional program induced by Aβ exposure, such as demonstrated by Prakash and Manchanda et al. with 5XFAD mice, and Haney et al. with AŴ peptide treatment.
Interestingly, we observed that prolonged exposure of APOE4 iMGLs to the ACSL1 inhibitor Triacsin C was cytotoxic and therefore unlikely to serve as a therapeutic agent. This is perhaps due to the importance of lipid droplets in buffering excess lipids associated with lipotoxicity. In fact, lipid droplets are thought to safeguard cells against various types of cellular stress (Jarc and Petan, 2019). Therefore, lipid accumulation influences many processes and must be fine-tuned rather than abrogated.
The new findings by the Wyss-Coray lab and the Davalos and Chopra labs offer exciting new mechanisms (i.e., inhibition of PI3 kinase or DGAT2) to alleviate lipid burden in microglia, but the long-term impact of these manipulations must be first carefully assessed.
Lin YT, Seo J, Gao F, Feldman HM, Wen HL, Penney J, Cam HP, Gjoneska E, Raja WK, Cheng J, Rueda R, Kritskiy O, Abdurrob F, Peng Z, Milo B, Yu CJ, Elmsaouri S, Dey D, Ko T, Yankner BA, Tsai LH. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron. 2018 Jun 27;98(6):1141-1154.e7. Epub 2018 May 31 PubMed.
Sienski G, Narayan P, Bonner JM, Kory N, Boland S, Arczewska AA, Ralvenius WT, Akay L, Lockshin E, He L, Milo B, Graziosi A, Baru V, Lewis CA, Kellis M, Sabatini DM, Tsai LH, Lindquist S. APOE4 disrupts intracellular lipid homeostasis in human iPSC-derived glia. Sci Transl Med. 2021 Mar 3;13(583) PubMed.
Victor MB, Leary N, Luna X, Meharena HS, Scannail AN, Bozzelli PL, Samaan G, Murdock MH, von Maydell D, Effenberger AH, Cerit O, Wen HL, Liu L, Welch G, Bonner M, Tsai LH. Lipid accumulation induced by APOE4 impairs microglial surveillance of neuronal-network activity. Cell Stem Cell. 2022 Aug 4;29(8):1197-1212.e8. PubMed.
Jarc E, Petan T. Lipid Droplets and the Management of Cellular Stress. Yale J Biol Med. 2019 Sep;92(3):435-452. Epub 2019 Sep 20 PubMed.
These interesting reports are a step forward. They suggest a functional link between ApoE and Aβ on the one hand, and phospho-tau, perhaps mediated by microglia-derived lipid droplets (LDs). It is possible that the light microscopic observation by Prakash et al. and Haney et al. of LDs in AD tissues are related to the extracellular droplets we observed intermingled with amyloid in tissues from a mouse model of AD (Leistner et al., 2003) and in post-mortem AD brain (Preprint: Gilbert et al., 2023). However, the size of the droplets observed by Prakash et al. and Haney et al. were 10- to 20-fold larger than those we observed by cryoET. This may be related to the resolution or sensitivity of light microscopy in detecting LDs. I suppose the choice of dyes to detect LDs is also important.
I wonder if the LDs described by these authors were located extracellularly, as they appeared to be in our cryoET studies. Extracellular droplets we observed by cryoET also appeared to possess one of several distinct structures, including spherical and cuboid droplets, and these were not observed in control brain. In contrast, LDs observed by light microscopy (Haney et al., Fig 2a) were also present in control brain, albeit with lower prevalence than in AD brain. It would be interesting to understand these differences and delineate the molecular signatures that might differentiate putative LD and extracellular droplet types.
Where is ApoE in all this? Is ApoE in close proximity to LDs, to extracellular droplets, or neither?
Leistner C, Wilkinson M, Burgess A, Lovatt M, Goodbody S, Xu Y, Deuchars S, Radford SE, Ranson NA, Frank RA. The in-tissue molecular architecture of β-amyloid pathology in the mammalian brain. Nat Commun. 2023 May 17;14(1):2833. PubMed.
Gilbert MA, Fatima N, Jenkins J, O'Sullivan TJ, Schertel A, Halfon Y, Morrema TH, Geibel M, Radford SE, Hoozemans JJ, Frank RA. In situ cryo-electron tomography of beta-amyloid and tau in post-mortem Alzheimer’s disease brain. 2023 Jul 18 10.1101/2023.07.17.549278 (version 1) bioRxiv.
These two papers support accumulating literature reporting dysregulation of microglial lipid metabolism in Alzheimer’s disease human brain and mouse models.
Prakash et al. showed LD formation in microglia in response to Aβ and corresponding increases in LDs in close proximity to plaques in human brain and in the 5xFAD mouse model of AD. Interestingly, LD formation was dependent on age and disease progression, and was prominent in the hippocampus, a pathologically vulnerable region in human and mouse brain. Prakash et al. showed that microglia with high LD load have deficits in phagocytosis, consistent with earlier findings that TREM2-deficient microglia failed to mount a functional immune response (Nugent et al., 2020). In that study, LD formation corresponded with an increase in cholesteryl ester formation and was shown to be dependent on apolipoprotein E4 (APOE) expression.
Similarly, Prakash et al. showed an increase in lipid storage in Aβ-treated microglia in the form of increased tricylglycerol and DGAT2 levels. This corresponded with reduced phagocytic function and free fatty acids. However, the analysis of lipidomic data was based on relative lipid levels, so it is unknown to what extent the absolute lipid content had changed. Interestingly, inhibition of DGAT2, and hence the lipid dyshomeostasis, rescued the deficit in phagocytosis. It is not clear if DGAT2 inhibition also rescued the lipidomic profile that led to the functional changes, because lipidomic data in response to DGAT2 inhibition were not provided. Due to the interconnected nature of lipid metabolism and signaling pathways, it will be necessary to determine the precise changes across the lipidome after DGAT2 inhibition to identify specific lipid species associated with amelioration of phagocytosis deficits.
Haney et al., identified an ACSL1-positive microglial state, which is most abundant in APOE4 carriers. They showed that fibrillar Aβ induction of triglyceride synthesis and of ACSL1 are dependent on APOE isoforms. This supports the critical role of APOE for mediating lipid storage and metabolism in response to pathological challenge. These authors further demonstrated that conditioned media from LD-rich microglia can induce tau phosphorylation and neurotoxicity. Further study is needed to identify the lipid and protein content of the secreted particles, be they exosomes, apolipoprotein particles, or other endogenous nanoparticle (eNP) culpable for these pathological effects.
One remaining challenge facing both studies is to account for the response of microglia functional states after acute isolation protocols. It is clear that microglia are sensitive to the physical and chemical environment, resulting in different activation states. It is not clear to what extent the isolated microglia represent activated or homeostatic states in situ. The homeostatic state of iPSC-derived microglia (iMG) is currently under investigation. However, it is demonstrated in these, and other, papers that there are differences among the responses of microglia isolated from different ages, sexes, and disease states. Further studies of microglia and cell-type-specific lipid dysregulation in situ, using emerging spatial transcriptomics and spatial mass spectrometry imaging technologies, are necessary to determine ground-truth, disease-relevant microglial deficits.
Further, the mechanism by which Aβ peptides, oligomers, and/or fibrils stimulate changes in lipid metabolism and microglial function remains unclear. Since different oligomeric and fibrillar Aβ species have been reported to have different functional activities, it will be important to clarify which specific oligomer or aggregate may differentially affect LD formation, phagocytosis, tau-phosphorylation, and neurotoxicity.
These studies highlight the association between lipid dysregulation and microglia dysfunction. This association remains correlative. The function of ApoE as a specific lipoprotein mediator of extracellular lipid transport and intracellular accumulation will need to be elucidated. Further mechanistic studies regarding the function of the dysregulated lipid species and their interactions with Aβ, and microglial function are critical for a full understanding of this interplay, which can help lead to more precise biomarkers and/or potential therapeutic targets in lipid metabolism.
Nugent AA, Lin K, van Lengerich B, Lianoglou S, Przybyla L, Davis SS, Llapashtica C, Wang J, Kim DJ, Xia D, Lucas A, Baskaran S, Haddick PC, Lenser M, Earr TK, Shi J, Dugas JC, Andreone BJ, Logan T, Solanoy HO, Chen H, Srivastava A, Poda SB, Sanchez PE, Watts RJ, Sandmann T, Astarita G, Lewcock JW, Monroe KM, Di Paolo G. TREM2 Regulates Microglial Cholesterol Metabolism upon Chronic Phagocytic Challenge. Neuron. 2020 Mar 4;105(5):837-854.e9. Epub 2020 Jan 2 PubMed.
To make a comment you must login or register.
To make an annotation you must Login or Register.
Copyright © 1996–2023 AlzForum Foundation Inc. All Rights Reserved.
- Call +91 8888006677 / 9422147673
- purchasesharp@gmail.com
- Wakad, Pimpri-Chinchwad, Maharashtra 411033